Reconciling contigs
Before running this step, you'll need to have completed the previous one (clustering contigs). I.e. you should have a Trycycler output directory (which I'll assume is called trycycler) with subdirectories for each of your good clusters, each of which contains a 1_contigs subdirectory. For convenience, you should probably also have deleted/renamed any subdirectories for bad clusters.
You'll also need the long reads you used in the previous step, which I'll assume are in reads.fastq.
Now that you have contig clusters to work on, Trycycler needs to reconcile the contigs within each cluster. This step is done per-cluster, so if your assemblies yielded three good contig clusters (e.g. one chromosome and two plasmids) then you will carry out this step on each of them.
Trycycler reconcile will:
- Perform an initial check to make sure the contigs look sufficiently similar to each other:
- relative lengths must be fairly close (e.g. one contig can't be twice as long as another)
- Mash distances must be quite small
- Ensure that all contig sequences are on the same strand
- If the replicon is circular:
- Fix any circularisation issues (i.e. add/remove sequence at each contig's start/end as necessary)
- Rotate the contigs to a common starting sequence
- Perform a final alignment check to make sure the normalised/circularised contigs are sufficiently similar to each other for the next step
If all goes well, this step will run and complete on its own. However, it may not and you'll need to manually intervene, for example deleting a contig sequence that is causing problems.
The Trycycler reconcile command must be run separately for each of your good clusters. Assuming your good clusters are numbers 1, 2 and 3, these are the commands you would run:
trycycler reconcile --reads reads.fastq --cluster_dir trycycler/cluster_001
trycycler reconcile --reads reads.fastq --cluster_dir trycycler/cluster_002
trycycler reconcile --reads reads.fastq --cluster_dir trycycler/cluster_003Trycycler reconcile should take a few minutes to complete – less for small contigs. Longer sequences and larger numbers of sequences will be slower, so a big bacterial chromosome with a lot of input contigs might take 10 minutes or more.
-
--linear: use this option if your input contigs are not circular. It will disable the circularisation-correction steps in Trycycler reconcile. -
--threads: this is how many threads Trycycler will use for read alignment. It will only affect the speed performance, so you'll probably want to use as many threads as you have available. -
--verbose: use this flag to display extra output. Mainly there for debugging purposes.
-
--max_mash_dist: if any of the sequences have a pairwise Mash distance of more than this (default = 0.02), then the contigs will fail the initial check. -
--max_length_diff: if any of the sequences have a pairwise relative length factor of more than this, then the contigs will fail the initial check. For example, if set to 1.1 (the default), then no contig can be more than 10% longer than any other.
-
--max_add_seqand--max_add_seq_percent: these control how much sequence Trycycler is willing to add to a contig to circularise it. For example, if they are set to 1000 and 5 (the defaults), then Trycycler will be willing to add up to 1000 bp or 5% of a contig's length (whichever is smaller) to circularise it. Any contig which requires more than 1000 bp or 5% of its length added to circularise will cause Trycycler reconcile to fail. -
--max_trim_seqand--max_trim_seq_percent: these control how much sequence Trycycler is willing to remove from a contig to circularise it. For example, if they are set to 50000 and 10 (the defaults), then Trycycler will be willing to remove up to 50000 bp or 10% of a contig's length (whichever is smaller) to circularise it. Any contig which requires more than 50000 bp or 10% of its length removed to circularise will cause Trycycler reconcile to fail.
-
--min_identity: if any of the sequences have a pairwise global alignment percent identity of less than this (default = 98), then the contigs will fail the final check. -
--min_1kbp_identity: if any of the sequences have a pairwise global alignment that drops below this percent identity (default = 25) in a 1-kbp sliding window, then the contigs will fail the final check.
When finished, Trycycler reconcile will make 2_all_seqs.fasta in the cluster directory, a multi-FASTA file containing each of the contigs ready for multiple sequence alignment.
Trycycler reconcile may not complete successfully, in which case you will have to intervene and run it again. Often this simply means excluding whichever contig is causing the problem (by deleting its FASTA file from the 1_contigs directory) and running Trycycler reconcile again with the remaining contigs. E.g. if you have eight input contigs for a cluster and one is preventing Trycycler reconcile from completing (for one of the reasons listed below) you can delete it and try again with the remaining seven contigs. Note that if you don't want to literally delete a contig (perhaps so you can change your mind later), it's sufficient to rename it so that it doesn't end with .fasta, e.g. changing A_contig_1.fasta to A_contig_1.fasta.bad.
Don't let this worry you – throwing out troublesome contigs at this step is normal. One of the reasons you prepared so many redundant assemblies was so you could have enough contigs in your clusters that losing one or two would not be a problem.
There are several reasons Trycycler reconcile might fail listed below, along with possible actions to take:
Ideally all the contig sequences will be approximately the same length. Some length variation is expected, due to the accumulation of small indels and/or circularisation problems (missing or duplicated sequence at the starts/ends). Trycycler reconcile will tolerate a bit of length difference, but if there is too much, it will refuse to continue. Similarly, if any of the pairwise Mash distances between contigs is too large, Trycycler reconcile will refuse to continue.
If this happens, the simplest solution is to just delete the offending contig(s) and run Trycycler reconcile again. This option is good if you have lots of input contigs so the loss of one or two won't be a problem. Alternatively, you can try to repair the offending contig(s). For example, if one contig is too long, you can try to fix it (remove the excess sequence) and try again. See Manually fixing overlap for suggestions on how this might work.
Length problems are especially common with Canu assemblies and small plasmids. In small plasmids, a modest amount of absolute overlap can create a large amount of relative overlap, and some assemblers will occasionally duplicate an entire small plasmid sequence in a single contig, making it twice as long as it should be.
Trycycler reconcile will try to circularise each contig using the other contigs as a reference. It does this by looking for the contig's start and end sequences in the other contig and then deciding if it needs to add or remove sequence to make it circular. If Trycycler reconcile cannot find the start/end sequences in other contigs or if it finds multiple instances of those sequences, it will not be able to continue.
How you proceed will depend on the reason for circularisation failure. Excluding the offending contig is an option, especially if you have a large number of contigs. If you don't have a lot of contigs and would therefore rather not discard anything you don't have to, then you can try to repair the circularisation. Sometimes a contig will contain junky sequence at its start/end, so trimming some sequence will allow Trycycler to continue. And sometimes adjusting the add_seq or trim_seq settings will allow Trycycler to continue.
Having an understanding of how Trycycler repairs circularisation may be of use here, so check out How circularisation repair works.
Trycycler reconcile performs a global alignment between all pairs of final sequences at the end of its execution. If any pair has a particularly bad pairwise identity (either overall or in a sliding window), that indicates a significant problem (e.g. a misassembly) with one of the sequences.
If this happens, you have two options. If one sequence in particular is causing the problem, it is probably best to delete that sequence and run Trycycler reconcile again. This is especially true if you have a good number of input contigs (e.g. six or more). Alternatively, if a large number of sequences are failing and/or you don't have very many input contigs, you can decrease the --min_identity and --min_1kbp_identity parameters to make Trycycler reconcile's final check more tolerant.
You can optionally run Trycycler dotplot on any problematic clusters to visualise the relationship between the sequences. For example:
trycycler dotplot --cluster_dir trycycler/cluster_002
This will create an image file (dotplots.png) in the cluster directory with a dotplot for all pairwise combinations of sequences. For example:
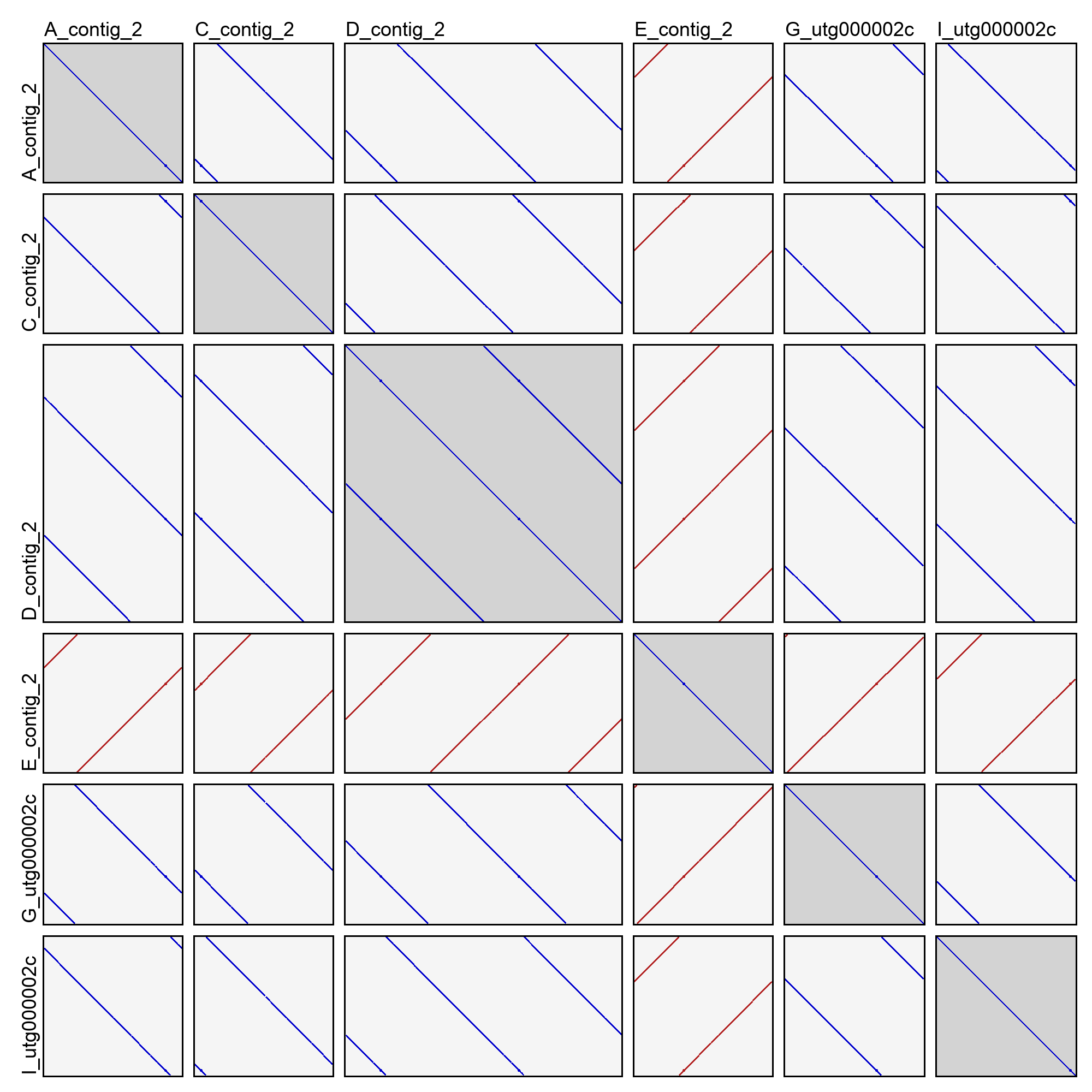
In the above example (taken from cluster 2 of the good demo dataset), you can see that most of the sequences are in very nice agreement with each other. They have shifted start positions relative to each other, but that's expected for contigs of a circular sequence. One of the contigs (E_contig_2) is on the opposite strand as the others, but that too is normal. Sequence D_contig_2, however, is a problem! It contains two entire copies of the same sequence, visible in the dotplot with itself and the dotplots with other sequences. It will need to be excluded or trimmed in order for reconciliation to succeed.
You should aim to have around four to eight contigs left after running Trycycler reconcile. Less than that (two or three) will not provide as many variants for the next steps and may affect your consensus sequence quality. More than that (nine or more) is fine but probably won't be of any extra benefit.
If you have too few contigs for your cluster, you might want to consider going back to the start of the pipeline and generating more input assemblies.
If you have plenty of contigs, you can delete some of the worst ones and run Trycycler reconcile again. Use the final check to guide you: delete the contigs with the lowest identities and largest indels relative to the other contigs.
For example, consider a Trycycler reconcile run where the final results look like this:
Overall pairwise identities:
A_contig_1: 100.000% 99.976% 99.978% 99.929% 99.978% 99.978% 99.978% 99.920% 99.979% 99.978%
B_contig_1: 99.976% 100.000% 99.979% 99.929% 99.978% 99.978% 99.978% 99.920% 99.979% 99.978%
C_contig_1: 99.978% 99.979% 100.000% 99.932% 99.979% 99.980% 99.980% 99.921% 99.981% 99.980%
D_contig_1: 99.929% 99.929% 99.932% 100.000% 99.931% 99.931% 99.931% 99.873% 99.932% 99.931%
E_contig_1: 99.978% 99.978% 99.979% 99.931% 100.000% 99.981% 99.980% 99.922% 99.981% 99.981%
F_utg000001c: 99.978% 99.978% 99.980% 99.931% 99.981% 100.000% 99.986% 99.929% 99.987% 99.987%
G_utg000001c: 99.978% 99.978% 99.980% 99.931% 99.980% 99.986% 100.000% 99.928% 99.987% 99.986%
H_utg000001c: 99.920% 99.920% 99.921% 99.873% 99.922% 99.929% 99.928% 100.000% 99.929% 99.928%
I_utg000001c: 99.979% 99.979% 99.981% 99.932% 99.981% 99.987% 99.987% 99.929% 100.000% 99.987%
J_utg000001c: 99.978% 99.978% 99.980% 99.931% 99.981% 99.987% 99.986% 99.928% 99.987% 100.000%
Worst-1kbp pairwise identities:
A_contig_1: 100.0% 99.6% 99.7% 48.0% 99.6% 99.4% 99.6% 38.9% 99.4% 99.6%
B_contig_1: 99.6% 100.0% 99.6% 48.0% 99.6% 99.5% 99.5% 39.0% 99.4% 99.6%
C_contig_1: 99.7% 99.6% 100.0% 48.0% 99.6% 99.4% 99.5% 39.0% 99.4% 99.6%
D_contig_1: 48.0% 48.0% 48.0% 100.0% 48.0% 48.0% 48.0% 39.0% 48.0% 48.0%
E_contig_1: 99.6% 99.6% 99.6% 48.0% 100.0% 99.6% 99.5% 39.0% 99.4% 99.5%
F_utg000001c: 99.4% 99.5% 99.4% 48.0% 99.6% 100.0% 99.5% 39.0% 99.4% 99.7%
G_utg000001c: 99.6% 99.5% 99.5% 48.0% 99.5% 99.5% 100.0% 39.0% 99.4% 99.6%
H_utg000001c: 38.9% 39.0% 39.0% 39.0% 39.0% 39.0% 39.0% 100.0% 39.0% 39.0%
I_utg000001c: 99.4% 99.4% 99.4% 48.0% 99.4% 99.4% 99.4% 39.0% 100.0% 99.4%
J_utg000001c: 99.6% 99.6% 99.6% 48.0% 99.5% 99.7% 99.6% 39.0% 99.4% 100.0%
Even though all sequences have technically passed (i.e. haven't failed the --min_identity and --min_1kbp_identity thresholds), two of them (D_contig_1 and H_utg000001c) are clearly worse than the rest. I would therefore exclude those two and run Trycycler reconcile again with the remaining eight contigs.