How read subsampling works
The Trycycler subsample command exists to turn a single long-read set into multiple subsets that can be used to generate input assemblies for Trycycler. Here are the steps in its operation:
The first thing Trycycler subsample does is randomly shuffle the reads. This is to account for any trends in the read set. For example, some Oxford Nanopore sequencing runs suffer from a drop in quality as the run progresses, so if your reads are in temporal order, the first n reads could be better than the last n reads. Randomly shuffling protects against this.
Trycycler subsample then decides what read depth each subset will be. This involves balancing two somewhat contradictory goals:
- Maximising the independence of the read subsets.
- Ensuring sufficient read depth in each subset for a good assembly.
These goals can be contradictory because higher subset read depths can help with assembly but decrease independence (due to more overlap between subsets). Conversely, lower subset read depths can make assembly harder but increase independence.
Trycycler subsample uses this function to find a read subset depth: y = m log2(4x/m)/2
- x is the full read set depth, y is the subsampled read depth and m is the minimum subset read depth
- The deeper your input reads, the deeper your read subsets will be (but only with logarithmic growth).
- Explore this function interactively on Desmos!
In plain language, this function says that an input read set of 25× depth will result in subsets of 25× depth (i.e. they'll be the same as the input set). It then requires a quadrupling of the input depth to add another 25× to the subsets. So 100× input depth gives 50× subset depth, 400x input depth gives 75× subset depth, 1600× input depth gives 100× subset depth, etc. The --min_read_depth option lets you change the value of m.
I chose these values because 25× is a good lower bound (a read set of this depth might assemble well), 50× is a good target (all assemblers seem to work well in this range) and anything over 100× is unlikely to significantly improve assemblies.
Trycycler cluster uses the mean read length to calculate how many reads should go in each subset to achieve the target subset read depth. This means the different subsets won't have the exact same number of bases, but they will be close.
To maximise the independence of the subsets, the starting read index is offset by r/c, where r is the read count and c is the subset count. The value of c is controlled by the --count option.
In these examples, the shuffled input reads are shown as a black bar at the top. The subsets are coloured bars, with their position indicating how much they overlap with each other. I've left --min_read_depth and --count at their default values (25 and 12, respectively) for each.
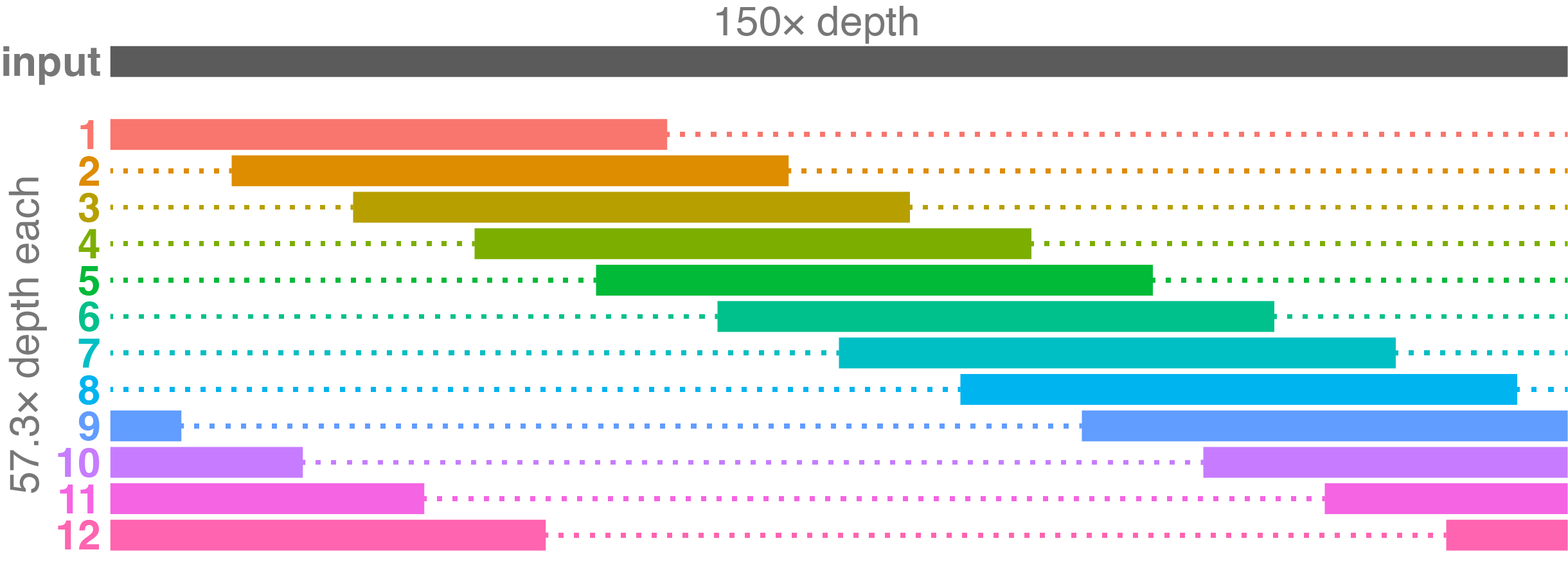
A 150× depth read set represents a typical case for Trycycler: not spectacular but not bad either. Each resulting subsampled set is completely independent from a few others but has some shared reads with most other sets. For example, read set 1 has no overlap with sets 6, 7, and 8, but it does overlap somewhat with the other sets.
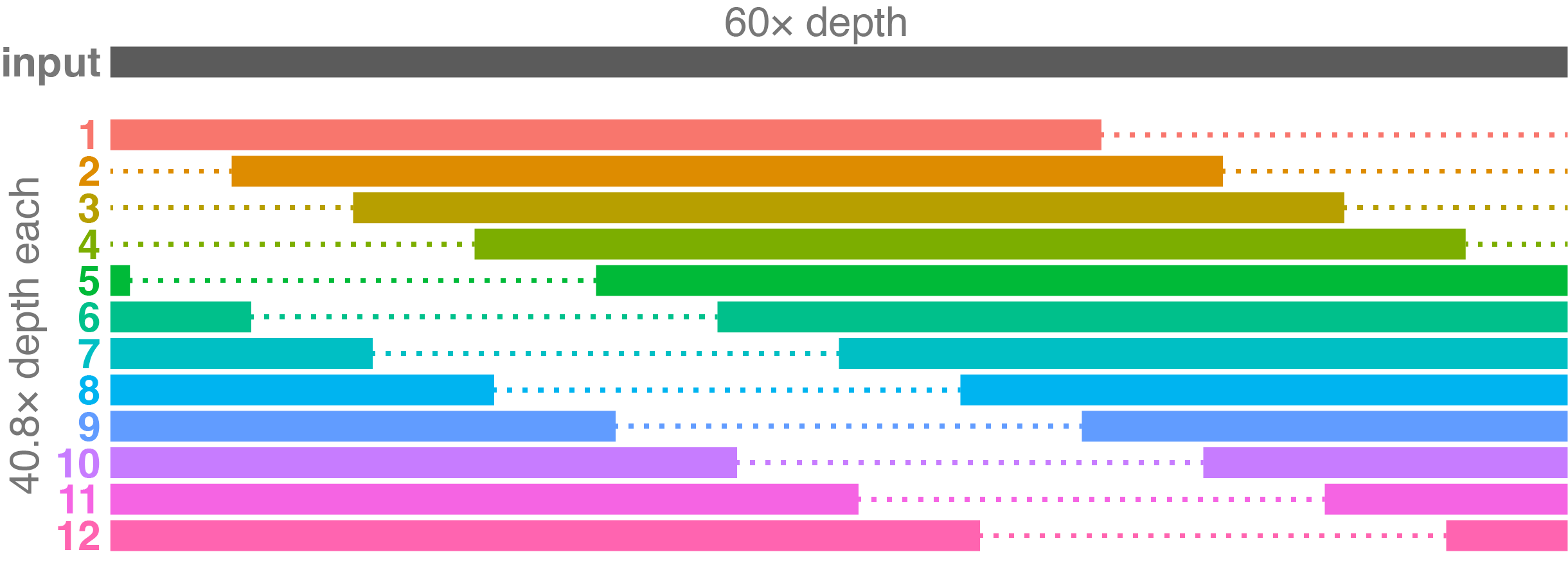
Since we have lower input depth in this example, Trycycler has made the subset depth lower as well. Unfortunately, the subsets overlap more, and no two subsets are completely independent.
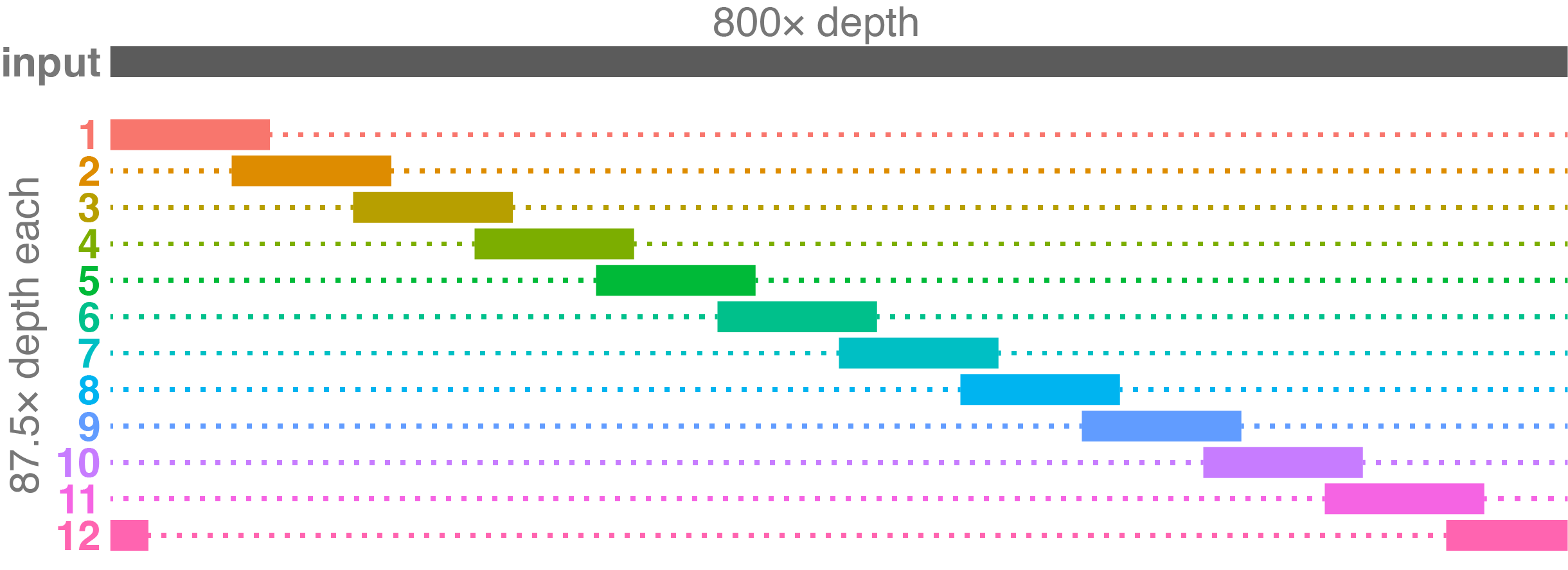
This example shows a case with a delightfully deep input set, giving us an ideal result. The subsets are deeper than in the the previous two examples, which might help them to assemble better. And they are quite independent, with each subset only sharing some overlap with two others.