需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量! 作业,理解测序reads,GC含量,质量值,接头,index,fastqc的全部报告,搜索中文教程,并发在论坛上面。
之前下载了所有的数据,但只有样本9 ~ 15才是mRNA-Seq测序结果,其中9-11为人类293个AKAP9敲除细胞,12-15为小鼠的AKAP9敲除细胞。也就是只要解压9 ~ 15就行了,即是SRR3589956 ~ SRR3589962.
先看一篇文章做过1000遍RNA-Seq的老司机告诉你如何翻车, 看一下事故现场,避开一些坑。
可以先用fastq-dump -h看一下帮助文件,分为如下几个部分:
- 输入: -A|--accession 序列号
- 处理中: Read Splitting, Full Spot Filters, Common Filters, Filters based on alignments, Filters for individual reads。 基本都是些过滤参数。不太常用
- 输出: -O|--outdir 输出文件夹, -Z|--stdout 输出到标准输出, --gzip/--bzip2 输出为压缩格式
- 多文件选项: 常用的就是--split-3
- 格式化: 分为序列, 质量等,不常用
所以基本上命令即使如下用法, 如果你觉得自己的空间够大就不需要用到--gzip参数。
for id in `seq 56 62`
do
fastq-dump --gzip --split-3 -O /mnt/f/Data/RNA-Seq -A SRR35899${id}
done
压缩的文件不要着急解压,有很多bash命令能够直接用于压缩文件,如zgrep,zcat,zless,zdiff等。
zcat SRR3589956_1.fastq.gz | head -n 4
@SRR3589956.1 D5VG2KN1:224:C4VAYACXX:5:1101:1159:2173 length=51
GGCGAGTGTAGGGCTGGCGCTGCCGGACGCGGTGCTAGTCGCCGGATGAAG
+SRR3589956.1 D5VG2KN1:224:C4VAYACXX:5:1101:1159:2173 length=51
B<BFBFBF0BFFFBFFBBFFIF<FFI<7<<BF<FFFFFFBB<BBBBBBBBB用这些z-tools能够节省大量磁盘空间。
高通量测序之所以能够能够达到如此高的通量的原因就是他把原来几十M,几百M,甚至几个G的基因组通过物理或化学的方式打算成几百bp的短序列,然后同时测序。

因为测序过程是边合成边测序(SBS),所以在建库的时候,短序列两段会加一些固定的碱基用于桥式PCR扩增,这些固定的碱基就是adapter(接头)。一般而言,还可以在接头加一些tag(index),用于标识这个read来自于哪个物种。目前的单细胞测序为了省钱,譬如10X genomic技术,都是在一个pool里面加多种接头。

在测序过程中,机器会对每次读取的结果赋予一个值,用于表明它有多大把握结果是对的。从理论上都是前面质量好,后面质量差。并且在某些GC比例高的区域,测序质量会大幅度降低。
因此,我们在正式的数据分析之前需要对分析结果进行质控。Jimmy大神就发帖专门指出”要充分了解你的测序数据--论QC的重要性“,http://www.biotrainee.com/thread-324-1-1.html 。
FASTQ文件每个序列通常为4行,分别为:
- Line 1 begins with a '@' character and is followed by a sequence identifier and an optional description (like a FASTA title line).
- Line 2 is the raw sequence letters.
- Line 3 begins with a '+' character and is optionally followed by the same sequence identifier (and any description) again.
- Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
FASTQ的文件示例:
@DJB775P1:248:D0MDGACXX:7:1202:12362:49613 1:Y:18:ATCACG
TGCTTACTCTGCGTTGATACCACTGCTTAGATCGGAAGAGCACACGTCTGAA
+
JJJJJIIJJJJJJHIHHHGHFFFFFFCEEEEEDBD?DDDDDDBDDDABDDCA
第一行序列名称 其中第一行的命名方式在v1.4后是 "@EAS139:\136:\FC706VJ:\2:\2104:\15343:\197393 1:\Y:\18:ATCACG"
| Tag | 描述 |
|---|---|
| DJB775P1 | the unique instrument name |
| 248 | the run id |
| D0MDGACXX | the flowcell id |
| 7 | flowcell lane |
| 1202 | tile number within the flowcell lane |
| 12362 | 'x'-coordinate of the cluster within the tile |
| 49613 | 'y'-coordinate of the cluster within the tile |
| 1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| Y | Y if the read is filtered, N otherwise |
| 18 | 0 when none of the control bits are on, otherwise it is an even number |
| ATCACG | index sequence |
在v1.4之前@HWUSI-EAS100R:6:73:941:1973#0/1
| Tag | 描述 |
|---|---|
| HWUSI-EAS100R | the unique instrument name |
| 6 | flowcell lane |
| 73 | tile number within the flowcell lane |
| 941 | 'x'-coordinate of the cluster within the tile |
| 1973 | 'y'-coordinate of the cluster within the tile |
| #0 | index number for a multiplexed sample (0 for no indexing) |
| /1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
** 第三行质量序列格式** 目前illumina使用的碱基质量格式为phred+33, 和Sanger的质量基本一致。
| Name | ASCII character range | Offset | Quality score type | Quality score range |
|---|---|---|---|---|
| Sanger, Illumina (versions 1.8 onward) | 33–126 | 33 | PHRED | 0–93 |
| Solexa, early Illumina (before 1.3) | 59–126 | 64 | Solexa | 5–62 |
| Illumina (versions 1.3–1.7) | 64–126 | 64 | PHRED | 0–62 |
不同版本的碱基质量Q和碱基错误率P的关系如下
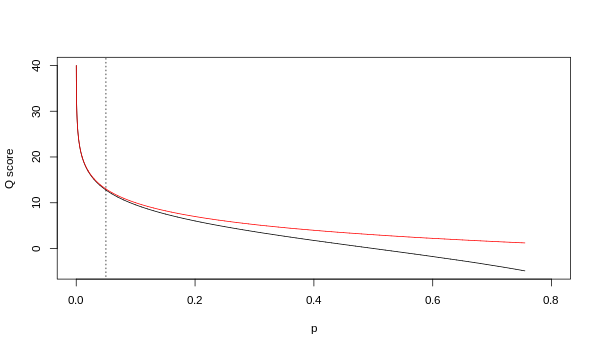
质量控制的软件很多,但是目前主要以fastqc为主。常见的用法:
fastqc seqfile1 seqfile2 .. seqfileN
常用参数:
-o: 输出路径
--extract: 输出文件是否需要自动解压 默认是--noextract
-t: 线程, 和电脑配置有关,每个线程需要250MB的内存
-c: 测序中可能会有污染, 比如说混入其他物种
-a: 接头
-q: 安静模式
FastQC有两种方式分析压缩的fastq文件
zcat SRR3589956_1.fastq.gz | fastqc -t 4 stdin
fastqc SRR3589956_1.fastq.gz
结果会得到一个html文件和一个zip压缩包。



具体含义可以看这里: http://jingyan.baidu.com/article/49711c6149e27dfa441b7c34.html
由于有14个结果,如果一个一个打开过去,一定会麻烦死,最好有一种一劳永逸的方法。 知乎的青山屋主写了一篇关于multiQC的教程(https://zhuanlan.zhihu.com/p/27646873, 介绍聚合多个QC结果进行演示的方法。
利用conda安装软件尤其简单,
conda install multiqc
multiqc --help
使用也很方便,
# 先获取QC结果
ls *gz | while read id; do fastqc -t 4 $id; done
# multiqc
multiqc *fastqc.zip --pdf
会有一个html文件用来了解总体情况

除了用multiQC查看多个QC结果以外,还可以专门写一个脚本看**每个样本的reads数量,GC含量,Q20,Q30的比例**
逻辑:
- 用python的zipfile模块打开zip文件,读取xx_data.txt的数据
- 读取每一行的数据,用正则表达式模块re,找到目标行
- 根据分隔符对每一行进行分割,进行赋值
- 由于只需要读取到>>Per base sequence quality pass这一部分,所以设置一个>>END_MODULE的计数器,数量超过2,就停止。
import re
import zipfile
# read the zip file
def zipReader(file):
qcfile = zipfile.ZipFile(file)
data_txt = [file for file in qcfile.namelist() if re.match(".*?_data\.txt", file)][0]
data = [bytes.decode(line) for line in qcfile.open(data_txt)]
return data
def fastqc_summary(data):
module_num = 0
bases = 0
Q20 = 0
Q30 = 0
for line in data:
if re.match('Filename', line):
filename = line.split(sep="\t")[1].strip()
if re.match('Total Sequence', line):
read = line.split(sep="\t")[1].strip()
if re.match('%GC', line):
GC = line.split(sep="\t")[1].strip()
if re.match("[^#](.*?\t){6}",line):
bases = bases + 1
if float(line.split("\t")[1]) > 30:
Q20 = Q20 + 1
Q30 = Q30 + 1
elif float(line.split("\t")[1]) > 20:
Q20 = Q20 + 1
if re.match(">>END", line) :
module_num = module_num + 1
if module_num >= 2:
break
Q20 = Q20 / bases
Q30 = Q30 / bases
summary = [filename, read, GC, str(Q20), str(Q30)]
return summary
if __name__ == '__main__':
import sys
for arg in range(1, len(sys.argv)):
data = zipReader(sys.argv[arg])
summary = fastqc_summary(data)
with open('summary.txt', 'a') as f:
f.write('\t'.join(summary) + '\n')